
Ross Chapman

Contact information
Chapman group: Genome stability and DNA repair mechanisms in cancer and genome diversification
Henry Wellcome Building of Genomic Medicine
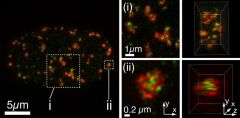 |
| BRCA1 and 53BP1 regulate DNA double strand break repair pathway choice. BRCA1 (green) and 53BP1 (red) enrich in mutually exclusive yet associated subnuclear volumes at sites of DNA damage as determined by 3D structured illumination super-resolution microscopy (OMX, Applied Precision). BRCA1 enrichment at DNA damage sites in S-phase (pictured) antagonizes 53BP1 association with chromatin to enable DSB repair by homologous recombination. |
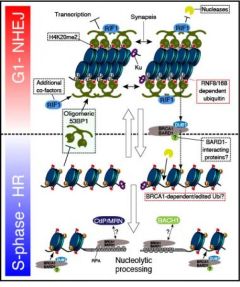 |
| Chromatin-dependent regulation of DNA repair. A dynamic competition between DNA repair factors and their residency in DNA damage-associated chromatin dictates DNA double-strand break repair pathway choice. |
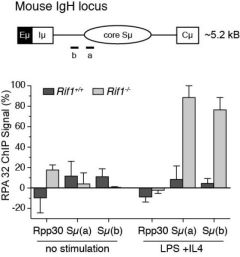 |
| RIF1-deficiency in mice results in aberrant nucleolytic processing of DSBs during immunoglobulin class-switch recombination. Chromatin immunoprecipitation of the single stranded DNA binding protein RPA32 at Immunoglobulin heavy chain (IgH) and control (Rpp30) loci in stimulated primary B-cells. |
Ross Chapman
Dr
Genome stability and DNA repair mechanisms in cancer and genome diversification
Research Summary: The accurate repair of DNA breaks is fundamental for protecting our genomes against cancer-causing mutations, however, the B and T lymphocytes of our immune systems deliberately induce and repair DNA breaks in a mutagenic fashion in order to adapt and diversify antigen receptor molecules. My group is interested in how cells and different tissues strike an appropriate equilibrium between accurate and mutagenic DNA repair mechanisms, so that we can understand why faults in this regulation lead to cancer, and devise innovative strategies to exploit these faults in cancer therapies.
DNA double-strand break repair in cancer and immunity: DNA double-strand breaks (DSBs) are highly toxic and must be accurately repaired to counteract the threat of human disease and oncogenic mutations. However, in some tissues mutagenic DSB repair is actually favoured, providing a molecular mechanism by which genetic material can be transferred between genetic loci to create diversity. To cope with this intrinsic discrepancy in desired DNA repair outcome between different cellular contexts, cells have evolved complex regulatory systems that maintain an appropriate equilibrium between competing DNA repair pathways, and that ensure DNA breaks are appropriately resolved.
Recent research from the group has shown that faults in a cell's ability to establish an appropriate equilibrium between accurate and mutagenic DSB repair pathways, links the mutagenic DNA repair systems that the developing immune system uses to diversify lymphocyte antigen receptor genes, to the mutational processes that triggers the onset of common cancers harbouring deficiencies in the homologous recombination (HR) DNA repair pathway. A particular focus of our research is to understand the molecular workings of a specialised branch of the non-homologous end joining (NHEJ) DSB repair pathway governed by the 53BP1 protein. In modelling the function of this mutagenic DNA repair pathway in developing and antigen-stimulated lymphocytes, we have discovered mechanisms that are required for the repair of DNA breaks during V(D)J recombination and immunoglobulin class-switch recombination (CSR). Our group has then gone on to demonstrate that the same processes are responsible for the mutations and genomic instability that accompanies mutation/loss of the tumour suppressor gene BRCA1 in hereditary breast and ovarian cancers. Given that poly-ADP ribose polymerase (PARP) inhibitors, modern therapeutics used in the treatment of BRCA-associated cancers, exploit these DNA repair defects to selectively kill cancer cells, our group has identified mechanisms in which these compounds act, and discovered drug-resistance mechanisms that may challenge their efficacy in the clinic.
Recent publications
CIP2A mediates mitotic recruitment of SLX4/MUS81/XPF to resolve replication stress-induced DNA lesions.
Journal article
de Haan L. et al, (2025), Nat Commun, 17
The ERCC6L2-MRI-KU complex coordinates NHEJ at staggered DNA double-strand breaks
Preprint
Reichl PI. et al, (2025)
Shieldin and CST co-orchestrate DNA polymerase-dependent tailed-end joining reactions independently of 53BP1-governed repair pathway choice
Journal article
CHAPMAN J., (2024), Nature Structural & Molecular Biology
BLM and BRCA1-BARD1 coordinate complementary mechanisms of joint DNA molecule resolution.
Journal article
Tsukada K. et al, (2024), Mol Cell, 84, 640 - 658.e10